

La UETeM se crea en el año 2003 bajo los auspicios de los Profesores Lado-Abeal y Araújo-Vilar en la Facultad de Medicina de Santiago de Compostela. Desde el año 2008 la unidad se dedica en exclusiva a la investigación en los aspectos clínicos y moleculares de los síndromes lipodistróficos infrecuentes. Desde este año, su director es el Dr.David Araújo-Vilar, Profesor Titular de Medicina y médico especialista en Endocrinología y Nutrición del Complexo Hospitalario Universitario de Santiago de Compostela (CHUS) (Figura 1), responsable de la Unidad de Lipodistrofias del servicio de Endocrinología del CHUS, que está integrada en el Centro de Referencia Nacional (CSUR) de Enfermedades Metabólicas Hereditarias y Centro Experto Europeo (MetabERN). Al mismo tiempo, el Dr.Araújo-Vilar es uno de los fundadores en 2012 del Consorcio Europeo de Lipodistrofias (ECLip, www.european-lipodystrophies.org/en). Desde 2012, los laboratorios de la UETeM están ubicados en el Centro de Investigación en Medicina Molecular y Enfermedades Crónicas (CiMUS) de la Universidad de Santiago de Compostela (Figura 2).

La UETeM está constituida en la actualidad por los siguientes miembros: David Araujo-Vilar (Endocrinólogo y Profesor Titular de Medicina), Joaquín Lado-Abeal (Endocrinólogo y Profesor Titular de Medicina, acreditado a catedrático de Universidad por la ANECA), Sofía Sánchez- Iglesias (doctora en Bioquímica y responsable de laboratorio), Cristina Guillín-Amarelle (endocrinóloga y doctora en Medicina), Antía Fernández-Pombo (médico residente de Endocrinología y Nutrición y estudiante de doctorado) y Yanis Bouzaher (graduado en Biología y estudiante de doctorado), colaborando también estudiantes de Medicina (María González-Alonso, Lia García-Formoso y Zeus Díaz-Vilela) (Figura 3 y 4).


Líneas de investigación 1.
La encefalopatía de Celia
1. La enfermedad: Esta enfermedad neurodegenerativa infantil fue descubierta en la UETeM en el año 2011 por el Dr Araújo-Vilar (Guillén-Navarro et al J Med Genet 2013). Es un trastorno muy infrecuente (menos de un caso por 1.000.000.000 de personas) caracterizado por una encefalopatía extremadamente grave que se inicia en la primera infancia. Sobre los 3 años estos niños sufren un involución neurológica rápida y progresiva que hace que pierdan sus funciones cerebrales (lenguaje, funciones motoras, interacción con el medio), apareciendo un cuadro de epilepsia mioclónica de muy difícil control, y que conduce al fallecimiento antes de los 9 años. En algunos casos (vide infra) además los niños presentan desde muy pequeños (meses) una pérdida pérdida generalizada de grasa ( l ipodist rofia) , lo que conl leva compl icaciones metaból icas y hepát icas (hipertrigliceridemia, diabetes, hígado graso). A día de hoy se han descrito 7 casos de la enfermedad, de los cuales 6 han fallecido. Este trastorno es una variante muy grave de la lipodistrofia congénita generalizada tipo 2 (síndrome de Berardinelli-Seip tipo 2). Recientemente, nuestro grupo ha identificado una variante de la encefalopatía de Celia debida a una mutación diferente en el gen BSCL2 (vide infra), con un cuadro clínico prácticamente superponible pero con una supervivencia algo mayor (10-11 años) (datos no publicados)
2. La causa: La encefalopatía de Celia, también denominada PELD (Progressive Encephalopathy with or without lipodystrophy, MIM: # 615924, https://www.omim.org/entry/ 615924?search=peld&highlight=peld) está causada por la mutación c.985C>T en el gen BSCL2, localizado en el cromosoma 11. Se trata de un trastorno recesivo, es decir, es necesario que ambos alelos del gen se encuentren afectados para que la enfermedad se desarrolle, siendo los padres portadores asintomáticos. Los pacientes homocigotos (es decir, los que tienen la mutación c.985C>T en los alelos materno y paterno) no presentan afectación del tejido adiposo, sin embargo los niños hetercigotos compuestos (la mutación c.985C>T está presente en uno de los alelos, y otra mutación diferente en el mismo gen lo está en el otro), además de la enfermedad neurodegenerativa presentan lipodistrofia generalizada. (Figura 5)
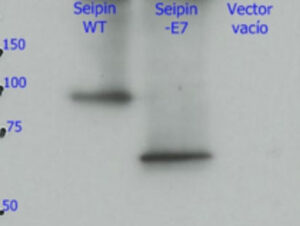
3. Los mecanismos: ¿Por qué esta mutación produce un daño tan grave en el cerebro? Estudios llevados a cabo en nuestro laboratorio (Guillén-Navarro et al J Med Genet 2013, Ruiz-Riquelme et al Neurobiol Dis 2015) han permitido desvelar porque la mutación c. 985C>T daña de una forma tan dramática el cerebro de estos niños. Este pequeño cambio en el exon 7 del gen BSCL2 provoca la desaparición del mismo (del exón 7) durante la transcripción (el paso de ADN a ARN). La pérdida de esta región del gen BSCL2 hace que la proteína que se traduce desde el ARN (la seipina) sea una proteína anormal o aberrante (le llamaremos seipina-Celia) con una tendencia a formar grandes agregados de seipina- Celia tanto en el citoplasma como el el núcleo de las neuronas. Estos macro-agregados de seipina-Celia no pueden ser eliminados por los mecanismos de defensa de las células cuando se encuentran con proteínas “enfermas” (lo que se llama UPR, respuesta a proteínas mal plegadas), lo que produce estrés del retículo endoplasmático, y llegado un punto, apoptosis (muerte cellular programada). Dicho de otra forma, a medida que pasan los meses, en las neuronas del cerebro de estos niños se acumulan grandes cantidades de la seipina-Celia lo que resulta “tóxico” para las mismas, por lo que se mueren. La pérdida constante de neuronas explica el cuadro neurológico progresivo y letal. (figura 6)
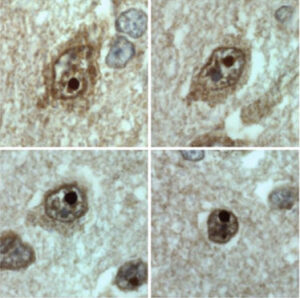
4. El tratamiento: A día de hoy no existe curación para esta enfermedad. No obstante, nuestro grupo ha hecho avances relevantes a la hora de enlentecer el daño neurológico. Desde hace 4 años y medio estamos tratando a la única niña viva con PELD con leptjna recombinante humana y una dieta rica en ácidos grasos poliinsaturados y suplementada con ácidos grasos omega-3. A día de hoy, esta niña, que ya cuenta con 8 años, no solo está viva sino que ha experimentado una modesta mejoría en los test psicométricos y, de forma llamativa, en los estudios de neuroimagen (Araujo-Vilar et al EJHG 2017, pendiente de aceptación). Nuestros estudios en modelos neuronales in vitro han confirmado que la asociación de leptina con ácido docosahexaenoico reduce la expresión de la seipina tóxica en un 30%. Estos resultados, aunque esperanzadores, deben ser tomados con enorme cautela ya que se trata de un único caso. En estos momentos, estamos valorando otras opciones terapéuticas sino para curar, por lo menos para retrasar el proceso.
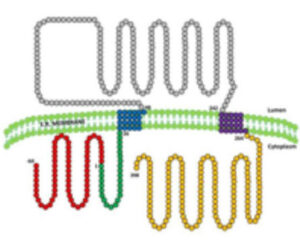 5. ¿Para que sirve la seipina?: La seipina es una proteína residente en el retículo endoplasmático de la que se conocen 3 isoformas (variantes) (figura 8). Estudios de nuestro grupo y de otros han demostrado que la isoforma más larga se expresa mayoritariamente en el sistema nervioso central, mientras que en los otros tejidos es la isoforma de tamaño intermedio la que predomina. Es muy probable que esta proteína tenga funciones diferentes según el tejido en el que se exprese. La función que mejor se ha estudiado está relacionada con la formación de gotas de lípidos en el tejido adiposo, de manera que la ausencia de seipina o la síntesis de una seipina defectuosa alteraría la formación de estas gotas que son cruciales en el desarrollo de los adipocitos. Pero además, existen datos que apuntan a que la seipina también juega un papel en el neurodesarrollo y nuestro grupo ha encontrado evidencias que sugieren que esta proteína podrían ejercer un papel protector frente al estrés oxidativo en la neuronas (artículo en preparación) (Figura 9).
5. ¿Para que sirve la seipina?: La seipina es una proteína residente en el retículo endoplasmático de la que se conocen 3 isoformas (variantes) (figura 8). Estudios de nuestro grupo y de otros han demostrado que la isoforma más larga se expresa mayoritariamente en el sistema nervioso central, mientras que en los otros tejidos es la isoforma de tamaño intermedio la que predomina. Es muy probable que esta proteína tenga funciones diferentes según el tejido en el que se exprese. La función que mejor se ha estudiado está relacionada con la formación de gotas de lípidos en el tejido adiposo, de manera que la ausencia de seipina o la síntesis de una seipina defectuosa alteraría la formación de estas gotas que son cruciales en el desarrollo de los adipocitos. Pero además, existen datos que apuntan a que la seipina también juega un papel en el neurodesarrollo y nuestro grupo ha encontrado evidencias que sugieren que esta proteína podrían ejercer un papel protector frente al estrés oxidativo en la neuronas (artículo en preparación) (Figura 9).
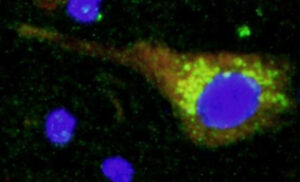
2. Búsqueda de nuevos genes implicados en la lipodistrofia parcial familiar. La lipodistrofia parcial familiar es una enfermedad mendeliana que puede ser causada por mutaciones en diferentes genes. Hasta la fecha se han identificado 7 genes, pero en más de un 50% de los pacientes no se han podido encontrar mutaciones en los mismos. Una de las lineas de investigación de nuestro grupo en encontrar nuevos genes responsable de este subtipo de lipodistrofias. Aunque a día de hoy hemos localizado un par de nuevos genes candidatos (artículos en preparación), nuestro proyecto más ambicioso en este campo lo estamos llegando a cabo en colaboración con la Universidad de Cambridge y el Welcome Trust Sanger Institute en el Reino Unido. Con ellos estamos analizado el genoma de más de 60 pacientes con lipodistrofia parcial familiar tipo 1.
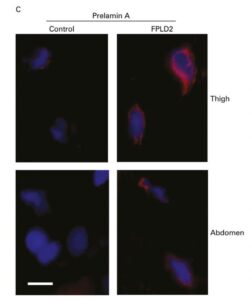 3. Mecanismos patogenéticos de la enfermedad de Dunnigan. La enfermedad de Dunnigan o Lipodistrofia Parcial Familiar tipo 2 es causada por ciertas mutaciones en el gen LMNA, un gen que codifica una proteína nuclear llamada Lamina A/C. A día de hoy no se sabe con certeza a través de qué mecanismos estas mutaciones dan lugar a una pérdida tan particular de tejido adiposo. Estudios llevados a cabo en nuestro laboratorio y por otros grupos sugieren que ciertas mutaciones en LMNA impiden un adecuado procesamiento de la Lamina A/C dando lugar a una acumulación de la forma inmadura de la proteína, la prelamina A. Este exceso de proteína inmadura alteraría los procesos de diferenciación adipocitaria (Araujo-Vilar et al. J Med Genet 2009) (Figura 10). No obstante, este mecanismo sería exclusivo de los adipocitos, porque en fibroblastos de estos pacientes dicha acumulación no tiene lugar (Y Tu et al. Nucleus 2016) (figura 11). Recientemente, en un trabajo en colaboración con Giovanna Lattanzi en Bolonia, hemos demostrado que la acumulación de prelamina A altera los procesos autofágicos, tanto en grasa blanca como en grasa parda en pacientes con esta enfermedad (C Pellegrini et al. Cell Death Differ. 2017, pendiente de aceptación). No obstante, recientemente, un trabajo del grupo de Philippe Collas en Noruega ha puesto de manifiesto la importancia de la alteración de ciertos mecanismos epigenéticos en el fallo de la adipogénesis en estos pacientes. En los próximos meses pretendemos iniciar una serie de experimentos en preadipocitos de pacientes con esta enfermedad para profundizar en las alteraciones epigenéticas.
3. Mecanismos patogenéticos de la enfermedad de Dunnigan. La enfermedad de Dunnigan o Lipodistrofia Parcial Familiar tipo 2 es causada por ciertas mutaciones en el gen LMNA, un gen que codifica una proteína nuclear llamada Lamina A/C. A día de hoy no se sabe con certeza a través de qué mecanismos estas mutaciones dan lugar a una pérdida tan particular de tejido adiposo. Estudios llevados a cabo en nuestro laboratorio y por otros grupos sugieren que ciertas mutaciones en LMNA impiden un adecuado procesamiento de la Lamina A/C dando lugar a una acumulación de la forma inmadura de la proteína, la prelamina A. Este exceso de proteína inmadura alteraría los procesos de diferenciación adipocitaria (Araujo-Vilar et al. J Med Genet 2009) (Figura 10). No obstante, este mecanismo sería exclusivo de los adipocitos, porque en fibroblastos de estos pacientes dicha acumulación no tiene lugar (Y Tu et al. Nucleus 2016) (figura 11). Recientemente, en un trabajo en colaboración con Giovanna Lattanzi en Bolonia, hemos demostrado que la acumulación de prelamina A altera los procesos autofágicos, tanto en grasa blanca como en grasa parda en pacientes con esta enfermedad (C Pellegrini et al. Cell Death Differ. 2017, pendiente de aceptación). No obstante, recientemente, un trabajo del grupo de Philippe Collas en Noruega ha puesto de manifiesto la importancia de la alteración de ciertos mecanismos epigenéticos en el fallo de la adipogénesis en estos pacientes. En los próximos meses pretendemos iniciar una serie de experimentos en preadipocitos de pacientes con esta enfermedad para profundizar en las alteraciones epigenéticas.
4. Nuevos tratamientos de las lipodistrofias infrecuentes
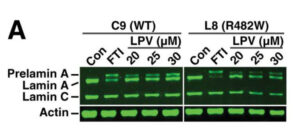
1. Leptina recombinante humana: A día de hoy, además de la dieta y el ejercicio físico, el tratamiento con leptina recombinante humana, es el único que permite un control adecuado de las complicaciones metabólicas y hepaticas asociadas a la lipodistrofia generalizada. Los datos de los que se dispone sobre su eficacia en la lipodistrofia parcial familiar sugieren que podría resultar beneficiosa en ciertos pacientes. La leptina no permite la restitución del tejido adiposo. La leptina recombinante humana (metreleptin) ha sido aprobada por las autoridades sanitarias en EEUU y Japón. En algunos países de Europa, entre ellos España, se lleva prescribiendo como uso compasivo desde hace 10 años. Nuestro grupo en Santiago de Compostela es el único en España que ha sido autorizado para su prescripción. A día de hoy estamos tratando a 11 pacientes con este fármaco, y estamos a la espera de autorización para tres pacientes más (Figura 12).
2. Otras alternativas terapéuticas:
1. Nuestro grupo participa en un ensayo clínico multicéntrico en fase 2/3 donde se está comprobando la eficacia de un nuevo tratamiento de la hipertrigliceridemia en la lipodistrofia parcial familiar. Se trata de un fármaco del tipo de los oligonucleótidos antisentido de segunda generación que se híbrida con el ARN mensajero del gen que codifica para la apoproteína apoC-III.
2. Simultáneamente, basándonos en experimentos ex vivo e in vitro llevados a cabo en nuestro laboratorio, disponemos de ciertas evidencias que nos hacen ser optimistas sobre ciertos fármacos que ya se están utilizando para otras patologías (reposicionamiento terapéutico) y que podrían ser de utilidad para al menos enlentecer el proceso neurodegenerativo de la encefalopatía de Celia. Esperamos que con el desarrollo de nuestro modelo murino transgénico podamos disponer de datos aún más sólidos al respecto.
 5. Registro Europeo de Lipodistrofias: Después de dos años de conversaciones y discusiones científicas, los miembros del Consorcio Europeo de Lipodistrofias (ECLip) (http:// www.european-lipodystrophies.org/en/) hemos aprobado el desarrollo de un registro europeo de pacientes con lipodistrofias infrecuentes (http://134.60.15.143:8080/login.xhtml) (Figura 13). El registro tendrá su sede en el universidad alemana de Ulm y será financiado con los fondos de los miembros de ECLip. La intención es mantener el registro un mínimo de 10 años y poder realizar un estudio pormenorizado de la historia natural de los diferentes subtipos de lipodistrofias (figura 14). En este sentido la participación de los pacientes y familiares será crítica. Según nuestras previsiones contamos con iniciar la recogida de datos de pacientes en noviembre de 2017.
5. Registro Europeo de Lipodistrofias: Después de dos años de conversaciones y discusiones científicas, los miembros del Consorcio Europeo de Lipodistrofias (ECLip) (http:// www.european-lipodystrophies.org/en/) hemos aprobado el desarrollo de un registro europeo de pacientes con lipodistrofias infrecuentes (http://134.60.15.143:8080/login.xhtml) (Figura 13). El registro tendrá su sede en el universidad alemana de Ulm y será financiado con los fondos de los miembros de ECLip. La intención es mantener el registro un mínimo de 10 años y poder realizar un estudio pormenorizado de la historia natural de los diferentes subtipos de lipodistrofias (figura 14). En este sentido la participación de los pacientes y familiares será crítica. Según nuestras previsiones contamos con iniciar la recogida de datos de pacientes en noviembre de 2017.

Producción científica (2003-2017)
1. Antía Fernández-Pombo, Pablo San Millán-Tejedor, Cristina Guillín-Amarelle, Ana I. Castro, Juan Carlos Guinarte-Cabada, Margarita Ventura-Victoria, David Araújo-Vilar. Morbid obesity and psychiatric disorders hiding a sex aneuploidy: a case of late diagnosis of 48,XXYY syndrome. Journal of Genetic Disorders & Genetic Reports, 2017 (pendiente de aceptación).
2. David Araújo-Vilar, Rosario Domingo-Jiménez, Álvaro Ruibal, Pablo Aguiar, Salvador Ibáñez-Micó, Miguel Garrido-Pumar, Miguel Ángel Martínez-Olmos, Concepción López-Soler, Cristina Guillín- Amarelle, María González-Rodríguez, Antonio Rodríguez-Núñez, Julián Álvarez-Escudero, Mercedes Liñares-Paz, Blanca González-Méndez, Silvia Rodríguez-García, Sofía Sánchez-Iglesias.The effects of metreleptin treatment and dietary intervention on neurological regression in Celia’s encephalopathy. European J Human Genet 2017 (pendiente de aceptación).
3. Camilla Pellegrini, Marta Columbaro, Sabino Prencipe, Davide Andrenacci, Laura Zanotti, Patricia Iozzo, Maria Angela Guzzardi, Cristina Capanni, Manuela Loi, Elisa Schena, David Araujo Vilar, Saverio Cinti, Paolo Morselli, Alessandra Gambineri, Giovanna Lattanzi. Altered adipocyte differentiation in FPLD2 is associated with unbalanced autophagy. In vitro and in vivo study of adipose tissue browning. Cell Death & Differentiation, 2017 (pendiente de aceptación).
4. Lado-Abeal J, Martinez-Sanchez N, Cocho JA, Martin-Pastor M, Castro-Piedras I, Saha AK, Lopez M. Septic shock effects on pig myocardial metabolites: a metabolomic approach. Scientific Report. 2017(pendiente de aceptación)
5. Cristina Guillín-Amarelle, Sofía Sánchez-Iglesias, Antonio Mera, Elena Pintos, Ana Castro-Pais, Leticia Rodríguez-Cañete, Julio Pardo, Felipe F. Casanueva, David Araújo-Vilar. Inflammatory myopathy in the context of a bizarre overlapping laminopathy. Archives of Endocrinology and Metabolism, 2017 (pendiente de aceptación)
6. Antía Fernández-Pombo, Javier A. Ossandon-Otero, Cristina Guillín-Amarelle, Sofía Sánchez-Iglesias, Ana I. Castro, Blanca González-Méndez, Silvia Rodríguez-García, Leticia Rodriguez-Cañete, Felipe F. Casanueva, David Araújo-Vilar. Bone mineral density in familial partial lipodystrophy. Clinical Endocrinology, 2017 (pendiente de aceptación).
7. Marta Giralt, Francesc Villarroya, David Araújo-Vilar. Lipodystrophies. Encyclopedia of Endocrine Diseases 2nd Edition, Elsevier 2017 (en prensa).
8. Chiquette E, Oral EA, Garg A, Araujo-Vilar D, Dhankhar P. Estimating the prevalence of generalized and partial lipodystrophy: findings and challenges. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy. 2017 (en prensa).
9. Jéru I, Vatier C, Araújo-Vilar D, Vigouroux C, Lascols O. Clinical Utility Gene Card for: Familial partial lipodystrophy. Eur J Hum Genet. 2017 Feb;25(2). doi: 10.1038/ejhg.2016.102.
10. Yiping Tu, Sofía Sánchez-Iglesias, David Araújo-Vilar, Loren G. Fong, Stephen G. Young. LMNA missense mutations causing familial partial lipodystrophy do not lead to an accumulation of prelamin A. Nucleus 2016 Sep 2;7(5):512-521.
11. Glynn N, Kenny H, Quisenberry L, Halsall DJ, Cook P, Tun T, McDermott J, Smith D, Thompson CJ, O’Gorman D, Boelen A, Lado-Abeal J, Agha A.. The effect of growth hormone replacement on the thyroid axis in patients with hypopituitarism: in vivo and ex vivo studies. Clinical Endocrinology. 86:747-754. 2017
12. Araújo-Vilar D, Barreiro J, Sánchez-Iglesias S, Guillín-Amarelle C. Acantosis nigricans in severe insulin resistance syndromes. An Pediatr (Barc). 2017 Mar;86(3):166-168. doi: 10.1016/j.anpedi.2016.01.003.
13. RJ Brown, D Araújo-Vilar, PT Cheung, D Dunger, A Garg, M Jack, L Mungai, EA Oral, N Patni, K Rother, J von Schnurbein, E Sorkina, T Stanley, C Vigouroux, M Wabitsch, R Williams, T Yorifuji. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab 2016 Dec;101(12):4500-4511.
14. Panikkath R, Panikkath D, Sánchez-Iglesias S, Araújo-Vilar D, Lado-Abeal J. An Uncommon Association of Familial Partial Lipodystrophy, Dilated Cardiomyopathy, and Conduction System Disease. J Investig Med High Impact Case Rep. 2016 Jul 15;4(3):2324709616658495.
15. Jéru I, Vatier C, Araújo-Vilar D, Vigouroux C, Lascols O. Clinical Utility Gene Card for: Congenital Generalized Lipodystrophy. Eur J Hum Genet. 2016 Nov;24(11). doi: 10.1038/ejhg.2016.53
16. C Guillín-Amarelle, S Sánchez-Iglesias, A Castro-Pais, L Rodriguez-Cañete, L Ordóñez-Mayán, Marcos Pazos, B González-Méndez, S Rodríguez-García, F.F. Casanueva, A Fernández-Marmiesse, D Araújo- Vilar. Type 1 Familial Partial Lipodystrophy: Understanding the Köbberling Syndrome. Endocrine, 2016 Nov;54(2):411-421, DOI: 10.1007/s12020-016-1002-x.
17. Sofía Sánchez-Iglesias, Alexander Unruh-Pinheiro, Cristina Guillín-Amarelle, Blanca González-Méndez, Alejandro Ruiz-Riquelme, Leticia Rodríguez-Cañete, Silvia Rodríguez-García, Encarnación Guillén- Navarro, Rosario Domingo-Jiménez, David Araújo-Vilar. Skipped BSCL2 transcript in Celia’s Encephalopathy (PELD): in vitro studies on senescence, adipogenesis and fatty acid treatment. PLOS ONE 2016 Jul 8;11(7):e0158874. doi: 10.1371/journal.pone.0158874.
18. Alejandro Ruiz-Riquelme, Sofía Sánchez-Iglesias, Alberto Rábano, Encarna Guillén-Navarro, Rosario Domingo-Jiménez, Adriana Ramos, Isaac Rosa, Peter Nilsson, Ángel García, David Araújo-Vilar, Jesús R. Requena. Larger aggregates of mutant seipin are part of the molecular basis of Celia’s Encephalopathy, a fatal neurodegenerative syndrome associated to the BSCL2 gene. Neurobiology of Diseases, 83:44-53, 2015.
19. Araujo-Vilar D, Sánchez-Iglesias S, Guillín-Amarelle C, Castro A, Lage M, Pazos M, Rial JM, Blasco J, Guillén-Navarro E, Domingo-Jiménez R, Del Campo MR, González-Méndez B, Casanueva FF. Recombinant human leptin treatment in genetic lipodystrophic syndromes: the long-term Spanish experience. Endocrine; 49(1):139-47, 2015.
20. Lado-Abeal J. Thyroid hormones are needed to sustain ”inappropriately” normal TSH during nonthyroidal illness syndrome: a clinical observation in severely ill patients with primary hypothyroidism. Neuroendocrinology Letters. 36 (1):101-107. 2015
21. Aráujo-Vilar D, Guillín-Amarelle C, Sánchez-Iglesias S, Castro A, Casanueva FF. Therapeutical uses of recombinant human leptin. Endocrinol Pediat,5 (Suppl): 27-42, 2014
22. Guillín-Amarelle C, Sánchez-Iglesias S, Araújo-Vilar D. Uncommon lipodystrophic syndromes. Medicina Clínica (Barc). 20;144(2):80-7, 2015
23. Guillén-Navarro E; Sánchez-Iglesias S, Domingo-Jimenez R; Victoria B; Ruiz-Riquelme A,Rábano A, Loidi L, Beiras A, González-Méndez B, Ramos A, Lopez González V, Ballesta-Martínez MJ, Garrido- Pumar M, Aguiar P, Ruibal A, Requena JR, Araújo-Vilar D. A new seipin-associated neurodegenerative syndrome. Journal of Medical Genetics. 50; 401-409, 2013
24. Castro I, Quisenberry L, Calvo RM, Obregon MJ, Lado-Abeal J. Lipopolysaccharide induced nonthyroidal illness syndrome causes hypothyroidism and conditions for reduced sensitivity to thyroid hormone.Journal of Molecular Endocrinology. 50: 255-266. 2013.
25. David Araújo-Vilar; Berta Victoria Martínez; Blanca González Méndez; Francisco Barreiro Morandeira; Beatriz Fernández Rodríguez; Rubén Cereijo; José Gallego Escuredo; Francesc Villarroya; Alberto Pañeda Menéndez. Histological and molecular features of lipomatous and non lipomatous adipose tissue from subjects with familial partial lipodystrophy due to LMNA mutations. Clinical Endocrinology. 76: 816-824, 2012.
26. Ana Ramos Levi; Angel Diaz Perez; Jose Manuel Cabezas Agricola; Maria Jesus Sobrido; Sergio Piñeiro; David Araújo-Vilar. Axonal neuropathy, long limbs and bumpy tongue: Think of MEN2B. Muscle&Nerve. 2012; 46: 961-964.
27. Joaquín Lado Abeal; R Albero Gamboa; David Araújo-Vilar; O Barca Mallo; Ignacio Bernabeu Morón; MT Calvo; Isabel Castro Piedras; J Martin Calamata; Fernando Palos Paz; Diego Peteiro. Clinical and molecular study of five families with resistance to thyroid hormones. Medicina Clínica (Barc). 2011; 137: 551 – 554.
28. Diego Peteiro-Gonzalez, Beatriz Fernandez-Rodriguez, Jose M Cabezas-Agrícola, David Araújo-Vilar. Severe localized lipoatrophy related to therapy with insulin analogs in type 1a diabetes mellitus. Diabetes Res Clin Pract, 91:e61-3, 2011.
29. Lado-Abeal J, Castro-Piedras I, Palos-Paz F, Labarta-Aizpún JI, Albero-Gamboa R. A Family with congenital hypothyroidism due to a combination of loss-of-function mutations in the TSH receptor (TSHR) and adenylate cyclase-stimulating G alpha-protein (GNAS) genes.
Thyroid.21:103-109. 2011
30. Berta Victoria, Marta Cuervo, José Manuel Cabezas-Agrícola, Blanca González-Méndez, Giovanna Lattanzi, Rosalba Del Coco, Lourdes Loidi, Francisco Barreiro, Carlos Calvo, David Araújo-Vilar. Reduced adipogenic gene expression in fibroblasts from a patient with type 2 congenital generalized lipodystrophy. Diabetic Medicine 27: 1178–1187, 2010
31. Lado-Abeal J, Romero A, Castro-Piedras I, Rodriguez-Perez A, Alvarez-Escudero J. Thyroid hormone receptors are down-regulated in skeletal muscle of patients with non-thyroidal illness syndrome secondary to non-septic shock.Eur J Endocrinol. 163:765-73. 2010.
32. Lado-Abeal J, Castro P, De la Calzada J, Castro-Piedras I, Celestino R, Espadinha C, Soares P, Sobrinho-Simoes M, Cameselle-Teijeiro J. Identification of a Paired Box Gene 8- Peroxisome Proliferator-Activated Receptor γ (PAX8-PPAR γ) rearrangement mosaicism in a patient with an autonomous functioning follicular thyroid carcinoma bearing an activating mutation in the TSH receptor. Endocrine Related Cancer. 17:599-610. 2010.
33. Peteiro D, Lee J, Rodriguez-Fontan J, Castro I, Cameselle-Teijerio JM, Beiras A, Alvarez CV, Hardy DM, Targovnik HM, Arvan P, Lado-Abeal J. New insights into thyroglobulin pathophysiology revealed by the study of a family with congenital goiter. Journal of Clinical Endocrinology and Metabolism. 95:3522-3526. 2010.
34. Joaquin Lado-Abeal, Rosa-Maria Calvo, Berta Victoria, Isabel Castro, Maria Jesus Obregon, David Araújo-Vilar. Regional decrease of subcutaneous adipose tissue in patients with type 2 familial partial lipodystrophy is associated with changes in thyroid hormone metabolism. Thyroid 20:419-24, 2010
35. Parajes S, Loidi L, Reisch N, Dhir V, Rose IT, Hampel R, Quinkler M, Conway GS, Castro-Feijóo L, Araújo-Vilar D, Pombo M, Dominguez F, Williams EL, Cole TR, Kirk JM, Kaminsky E, Rumsby G, Arlt W, Krone N. Functional Consequences of Seven Novel Mutations in the CYP11B1 Gene: Four Mutations Associated with Nonclassic and Three Mutations Causing Classic 11{beta}-Hydroxylase Deficiency. J Clin Endocrinol Metab. 95:779-88, 2010
36. Araújo-Vilar D, Giovanna Lattanzi, Blanca González-Méndez, Ana Teresa Costa-Freitas, Daniel Prieto, Marta Columbaro, Elisabetta Mattioli, Berta Victoria, Noelia Martínez-Sánchez, Alia Ramazanova, Máximo Fraga, Andrés Beiras, Jerónimo Forteza, Lourdes Domínguez-Gerpe, Carlos Calvo, Joaquin Lado-Abeal Site-dependent differences in both prelamin A and adipogenic genes in subcutaneous adipose tissue of patients with type 2 familial partial lipodystrophy. J Medical Genetics 46: 40-8, 2009
37. Lago, R, Iglesias MJ, San-Jose, E, Areal C, Eiras A, Araujo-Vilar D, Lado-Abeal J. Dominguez-Gerpe L. Prevalence and functional analysis of the S107P SNP (rs6647476) of the SLC16A2 gene in the male population of Northwest Spain (Galicia). Clinical Endocrinology 70(4):636-43, 2009
38. Castro-Piedras I, Lima L, Seoane R, Lado-Abeal J. Identification and functional characterization of two novel activating TSH receptors mutants in toxic thyroid follicular adenomas. Thyroid.19: 645-649. 2009.
39. Domínguez-Gerpe L, Araújo-Vilar D. Prematurely Aged Children: Molecular Alterations Leading to Hutchinson-Gilford Progeria and Werner Syndromes. Current Aging Science 1:202-212, 2008
40. Palos-Paz F, Perez-Guerra O, Cameselle-Teijeiro J, Rueda-Chimeno C, Barreiro-Morandeira F, Lado- Abeal J and the Galician Group for the Study of Toxic Multinodular Goitre: Araujo-Vilar D, Argueso R, Barca O, Botana M, Cabezas-Agrícola JM, Catalina P, Dominguez Gerpe L, Fernandez T, Mato A, Nuño A, Penin M, Victoria B. Prevalence of mutations in TSHR, GNAS, PRKAR1A and RAS genes in a large series of toxic thyroid adenomas from Galicia, an iodine deficient area in NW Spain. European J Endocrinol 159: 623-31, 2008
41. Eva Fernández-Rodríguez, Rocío Villar-Taibo, Iria Pinal-Osorio, José Manuel Cabezas-Agrícola, Urbano Anido-Herranz, Alma Prieto, Felipe F Casanueva, David Araujo-Vilar. Severe hypertension and hypokalemia as first clinical manifestations in ectopic Cushing’s syndrome. Arquivos Brasileiros de Endocrinologia & Metabologia, 52(6):1066-70, 2008
42. Araújo-Vilar D, Sarmiento LM, Barros N, Ana Costa da Silva-Freitas, Alia Ramazanova, Joaquín Lado- Abeal, Carlos Calvo, F. Palos and Lourdes Dominguez. Resting metabolic rate is reduced in some obese subjects’ subpopulations. Obesity and Metabolism 4: 22-27, 2008
43. Araújo-Vilar D, Joaquin Lado-Abeal, Fernando Palos-Paz, Giovanna Lattanzi, Manuel Angel Bandín, Diego Bellido, Lourdes Domínguez-Gerpe, Carlos Calvo, Oscar Pérez, Alia Ramazanova, Noelia Martínez-Sánchez, Berta Victoria, Costa Freitas AT. A new clinical condition associated with a novel mutation in LMNA gene, characterized by partial lipodystrophy, insulin-resistance, aortic stenosis and hypertrophic miocardiopathy. Clinical Endocrinology 69(1):61-8, 2008.
44. Fernando Palos, María ER García-Rendueles, David Araujo-Vilar, Maria Jesús Obregon, Rosa Maria Calvo, Jose Cameselle-Teijeiro, Susana B Bravo, Oscar Perez-Guerra, Lourdes Loidi, Barbara Czarnocka, Paula Alvarez, Samuel Refetoff, Lourdes Dominguez-Gerpe, Clara V Alvarez, Joaquin LadoAbeal. Pendred syndrome in two Galician families: insights into clinical phenotypes through cellular, genetic and molecular studies. J Clin Endocrinol Metab. 93(1):267-77, 2008.
45. Palos-Paz F, Perez-Guerra O, Cameselle-Teijeiro J, Rueda-Chimeno C, Barreiro-Morandeira F, Lado- Abeal J (and the Galician Group for the Study of Toxic Multinodular Goitre). Prevalence of mutations in TSHR, GNAS, PRKAR1A and RAS genes in a large series of toxic thyroid adenomas from Galicia, an iodine deficient area in NW Spain. European Journal of Endocrinology. 159:623-631. 2008.
46. Rodriguez-Perez A, Palos-Paz F, Kaptein E, Visser TJ, Dominguez-Gerpe L, Alvarez-Escudero J, Lado- Abeal J. Identification of Molecular Mechanisms Related to Non-Thyroidal Illness Syndrome in Skeletal Muscle and Adipose Tissue from Patients with Septic Shock. Clinical Endocrinology (Oxf). 68:821-827. 2008.
47. J.F. Ascaso, E. Aguillo, D. Araujo-Vilar et al. Diabetes mellitus y riesgo cardiovascular. Recomendaciones del grupo de trabajo Diabetes mellitus y enfermedad cardiovascular de la Sociedad Española de Diabetes 2006. Clin Invest Arterioscl. 19(3):167-72, 2007
48. Lado-Abeal J, Lorenzo-Solar M, Lago-Lestón R, Palos-Paz F, Domingez-Gerpe L. Hyperglycaemic hyperosmolar non-ketotic state as a cause of low gonadotropin levels in postmenopausal diabetic women: a role for severe hypernatraemia. Journal of Neuroendocrinology. 19:983-987. 2007
49. Villamil Cajoto I, Araujo-Vilar, D. Tratamiento con vitamina D en la infancia: discusión de la evidencia. An. Med. Int. 23:446-48, 2006
50. Araujo-Vilar D, Grupo De Trabajo Diabetes Mellitus y Enfermedad cardiovascular. Documento de consenso: Diabetes mellitus y riesgo cardiovascular. Av. Diabetol 22: 143-48, 2006
51. Loidi L, Quinteiro C, Parajes S, Barreiro J, Leston DG, Cabezas-Agricola JM, Sueiro AM, Araujo-Vilar D, Castro-Feijoo L, Costas J, Pombo M, Dominguez F. High variability in CYP21A2 mutated alleles in Spanish 21-hydroxylase deficiency patients, six novel mutations and a founder effect. Clin Endocrinol (Oxf). 2006 Mar;64(3):330-6, 2006.
52. Dumitrescu AD, Liao XH, Abdullah SYM, Lado-Abeal J, AbdulMajed F, Moeller LC, Boran G, Schomburg L, Weiss RE, Refetoff S. Mutations in the selenocystein insertion sequence binding protein (SBP)2 produce a thyroid phenotype. Nature Genetics. 37:1247-1252. 2005
53. Lado-Abeal J, Dumitrescu AM, Cohen RN, Pohlenz J, Liao XH, Lebrethon MC, Verloes A, Refetoff S.A de novo mutation in an Already Mutant Nucleotide of the Thyroid Hormone Receptor Beta Gene perpetuates resistance to thyroid hormone. Journal of Clinical Endocrinology and Metabolism. 90:1760-1767. 2005.
54. Lado-Abeal J, Robert-McComb JJ, Qian XP, Leproult R, Van Cauter E, Norman RL. Gender differences in the neuroendocrine response to short-term energy restriction in non-human primates. Journal of Neuroendocrinology. 17:435-44. 2005.
55. Graña Barcia M, Liz Leston JL, Lado Abeal J. Subcutaneous administration of pulsatile GnRH decreases serum FSH and LH levels in women with polycystic ovary syndrome: a preliminary study. Fertility and Sterility. 83:1466-1472. 2005
56. García-Estevez DA, Araujo-Vilar D, Saavedra-Gonzalez A, Fiestras-Janeiro G, Cabezas-Cerrato J. Analysis of the relationship between body mass index, insulin resistance, and beta-cell function: a crosssectional study using the minimal model. Metabolism. 53(11):1462-6, 2004.
57. Lado-Abeal J, DeValk E, Pacini F, Refetoff S. Study of the effect of recombinant human thyroid stimulating hormone on Leydig cells function in men with differentiated thyroid carcinoma. Thyroid. 13:649-652. 2003.
58. Narendran P, Lado-Abeal J, Moeller LC, Refetoff S. Partial thyroxine-binding globulin (TBG) deficiency in a family with no detectable mutation of the TBG gene. Clinical Endocrinology. 59:823-825. 2003.
59. Cabezas-Cerrato, Araújo-Vilar D. Resistencia a la acción de la insulina. Evolución histórica del concepto. Técnicas para el estudio in vivo en humanos. Endocrinología y Nutrición. 50: 396-406, 2003
60. Garcia-Estevez DA, Araujo-Vilar D, Fiestras-Janeiro G, Saavedra-Gonzalez A, Cabezas-Cerrato J. Insulin resistance in essential hypertension: a conflictive point of view. Diabet Med. 2003 Dec;20(12): 1035, 2003.
61. Araujo-Vilar D, Loidi L, Dominguez F, Cabezas-Cerrato J. Phenotypic gender differences in subjects with familial partial lipodystrophy (Dunnigan variety) due to a nuclear lamin A/C R482W mutation. Horm Metab Res. 2003 Jan;35(1):29-35, 2003.
62. Garcia-Estevez DA, Araujo-Vilar D, Fiestras-Janeiro G, Saavedra-Gonzalez A, Cabezas-Cerrato J. Comparison of several insulin sensitivity indices derived from basal plasma insulin and glucose levels with minimal model indices. Horm Metab Res. 35(1):13-7, 2003.
63. Araújo-Vilar D. Lipodistrofias: bases moleculares y características clínicas. Endocrinología y Nutrición. 50: 133-44, 2003
Artículos en preparación:
1. David Araújo-Vilar, Ferruccio Santini. Diagnosis and treatment of lipodystrophies: a step-by-step approach.
2. D Araujo-Vilar et al. A case of Celia’s encephalopathy due to a new BSCL2 mutation.
3. S. Sánchez-Iglesias, D Araujo-Vilar et al. Role of BSCL2 transcripts in human brain and its relationships with ROS protection and peroxisomes generation.
4. Cristina Guillín-Amarelle, Sofía Sánchez-Iglesias, David Araújo-Vilar. VARIABLE EXPRESSIVITY IN LMNA-ASSOCIATED FAMILIAL PARTIAL LIPODYSTROPHY.
5. Sofía Sánchez-Iglesias, Marcos M. Lima-Martínez, Cristina Guillín-Amarelle, S, David Araújo-Vilar. LEVOTHYROXINE TREATMENT IN RABSON-MENDENHALL SYNDROME.
6. C Guillín-Amarelle, S Sánchez-Iglesias, A Fernández-Pombo, D Araujo-Vilar. How to diagnose a LMNAassociate lipodystrophy. Nucleus 2017
Organización de congresos y otras actividades científicas
Reuniones del Consorcio Europeo de Lipodistrofias:
• Bolonia, 10-11 Febrero 2014 (Prof. Giovanna Lattanzi)-Acta fundacional. (figura 15)

• Paris, 30 Abril 2015 (Prof. Corinne Vigouroux) (Figura 16)

• Santiago de Compostela, 8-9 Marzo 2016 (Prof. David Araujo-Vilar) (Figura 17)

• Roma, 21-22 Abril 2017 (Prof. Giuseppe Novelli) (Figura 18, 19, 20)



• Ulm, 7-8 Julio 2017 (Prof. Martin Wabitsch)
• Münster, 27-28 Abril 2018 (Prof. Hartmut Schmidt)
Colaboraciones con otros grupos de investigación:
• Dra Rosario Domingo, Unidad de Neuropediatría, HospitalUniversitario Virgen de la Arrixaca, Murcia
• Prof. Giovanna Lattanzi, National Research Council of Italy – Institute for Molecular Genetics, Bolonia, Italia.
• Prof. Corinne Vigouroux, Centre de Recherche Saint-Antoine INSERM UMR_S 938. Faculté de Médecine Pierre et Marie Curie (Figura 21) .

• Dr. David Savage, National Severe Insulin Resistance Service. Wellcome Trust-MRC Institute of Metabolic Science. Cambridge University Hospital NHS Foundation Trust . Addenbrookes Hospital. UK (Figura 22).

• Prof. Steve Young, University of California, Los Ángeles, EEUU.
• Dra. Rebecca Brown, National Institute of Diabetes and Digestive and Kidney, Diseases( NIDDK), Diabetes, Endocrinology, and Obesity Branch (DEOB), Bethesda, MD, EEUU.
• Prof. Francesc Villarroya, Departamento de Bioquímica y Biología Molecular, Universitat de Barcelona (Figura 23).

• Prof. Martin Wabitsch, Pediatrics and Adolescent Medicine, University Medical Center Ulm, Alemania.
• Dra. Margarita Lopez Trascasa, Servicio de Inmunología, Hospital Universitario de La Paz, Madrid.
• Prof. Elif A. Oral, Universidad de Michigan, EEUU (Figura 24)

Contacto
1. Para asuntos relacionados con proyectos de investigación:
Prof. David Araujo-Vilar
UETeM, Lab 3, Planta 2
CIMUS, Avda Barcelona 3
15707 Santiago de Compostela
Tfn: +34 639 393 458
Mail: david.araujo@usc.es; lipodistrofias@gmail.com
2. Para asuntos médicos:
Prof. David Araujo-Vilar
Servicio de Endocrinología y Nutrición
Complexo Hospitalario Universitario de Santiago.
(Hospital Médico-Cirúrxico de Conxo, Rua de Ramón Baltar s/n, 15782 Santiago)
Mail: david.araujo@usc.es; lipodistrofias@gmail.com
Nota: La asistencia sanitaria en un hospital público es gratuita. Al ser nuestro servicio unidad de referencia en España para Lipodistrofias, es recomendable que aquellos pacientes que no pertenezcan al Servicio Galego de Saude (SERGAS) soliciten a su médico de cabecera o especialista que solicite ASISTENCIA SANITARIA a través del sistema SIFCO (Sistema de Información del Fondo de Cohesión) para que puedan ser atendidos en nuestro servicio.